全部
▼
搜索
熱搜:
位置:中冶有色 >
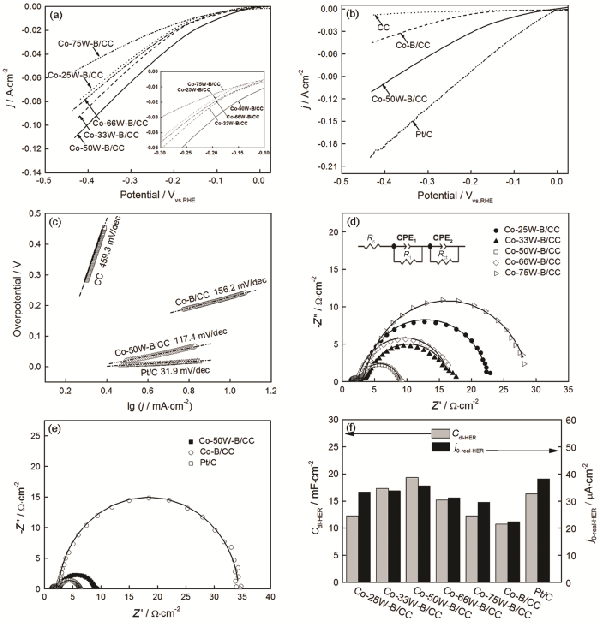
> 非晶Co-W-B/碳布復(fù)合電極材料的制備及其電解水催化性能
 924
編輯:中冶有色技術(shù)網(wǎng)
來源:施嘉倫,盛敏奇,吳瓊,呂凡
924
編輯:中冶有色技術(shù)網(wǎng)
來源:施嘉倫,盛敏奇,吳瓊,呂凡





 分享 0
分享 0
 舉報(bào) 0
舉報(bào) 0
 收藏 0
收藏 0
 反對(duì) 0
反對(duì) 0
 點(diǎn)贊 0
點(diǎn)贊 0

 中冶有色技術(shù)平臺(tái)
中冶有色技術(shù)平臺(tái)合材料制備/加工第八屆學(xué)術(shù)會(huì)議")

 2024年12月27日 ~ 29日
2024年12月27日 ~ 29日 合材料應(yīng)用技術(shù)創(chuàng)新論壇") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 同創(chuàng)新發(fā)展論壇") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 廢水處理與資源化利用交流會(huì)") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 粒礦物選礦技術(shù)大會(huì)") 2025年03月25日 ~ 27日
2025年03月25日 ~ 27日 
 京公網(wǎng)安備 11010702002294號(hào)
京公網(wǎng)安備 11010702002294號(hào)










































































