全部
▼
搜索
熱搜:
位置:中冶有色 >
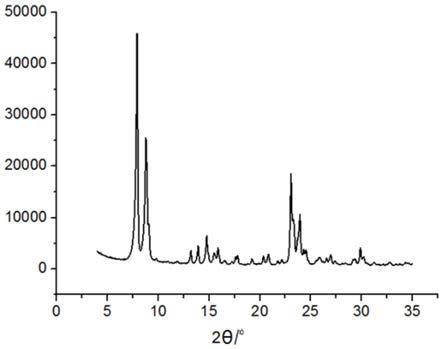
> MFI拓?fù)鋵W(xué)結(jié)構(gòu)硅分子篩及其制備方法和應(yīng)用與流程
 418
編輯:中冶有色技術(shù)網(wǎng)
來(lái)源:浙江恒瀾科技有限公司
418
編輯:中冶有色技術(shù)網(wǎng)
來(lái)源:浙江恒瀾科技有限公司
 分享 0
分享 0
 舉報(bào) 0
舉報(bào) 0
 收藏 0
收藏 0
 反對(duì) 0
反對(duì) 0
 點(diǎn)贊 0
點(diǎn)贊 0

 中冶有色技術(shù)平臺(tái)
中冶有色技術(shù)平臺(tái)合材料制備/加工第八屆學(xué)術(shù)會(huì)議")

 2024年12月27日 ~ 29日
2024年12月27日 ~ 29日 三稀礦產(chǎn)資源技術(shù)交流會(huì)") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 碳纖維與復(fù)合材料應(yīng)用技術(shù)創(chuàng)新論壇") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 廢水處理與資源化利用交流會(huì)") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 資源科技創(chuàng)新發(fā)展論壇") 2025年04月27日 ~ 29日
2025年04月27日 ~ 29日 
 京公網(wǎng)安備 11010702002294號(hào)
京公網(wǎng)安備 11010702002294號(hào)










































































