全部
▼
搜索
熱搜:
位置:中冶有色 >
> 氧氣DBD等離子體處理PBO纖維表面及其對雙馬樹脂基復(fù)合材料界面性能的影響
 920
編輯:中冶有色技術(shù)網(wǎng)
來源:劉哲,陳博涵,陳平
920
編輯:中冶有色技術(shù)網(wǎng)
來源:劉哲,陳博涵,陳平

| Properties | Values |
|---|---|
| Viscosity/Pa·s(80℃) | 0.5~1.5 |
| Softening point/℃ | 20~30 |
| Tensile strength/MPa | 65 |
| Tensile modulus/GPa | 3.3 |
| Elongation at break/% | 2.5 |
| Fracture energy release rate/J·m-2 | 298 |
| Tg/℃ |
According to E’:240 According to tanδ:270 |

| Parameter | Temperature | Pressure | Time |
|---|---|---|---|
| Preheat | Room temperature~125℃ | 0 MPa | 30 min |
| Gel | 125℃ | 0.5 MPa | 50~60 min |
| Solidify | 190℃ | 1.5 MPa | 3 h |
| Post curing | 235℃ | 1.5 MPa | 3 h |
| Cooling | Natural cooling to room temperature |

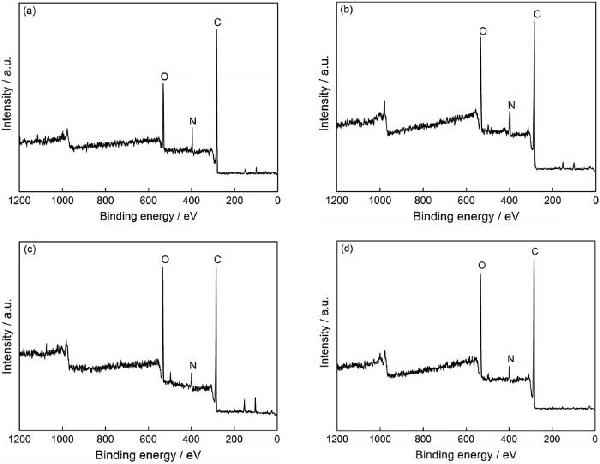
|
Treatment time/s |
Relative elemental concentration/%, atomic fraction | Atomic ratio | |||
|---|---|---|---|---|---|
| C | O | N | O/C | N/C | |
| 0 | 77.22 | 18.01 | 4.77 | 0.23 | 0.06 |
| 12 | 74.20 | 22.14 | 4.66 | 0.30 | 0.06 |
| 24 | 70.97 | 24.83 | 4.21 | 0.35 | 0.06 |
| 36 | 76.84 | 20.50 | 2.66 | 0.27 | 0.03 |
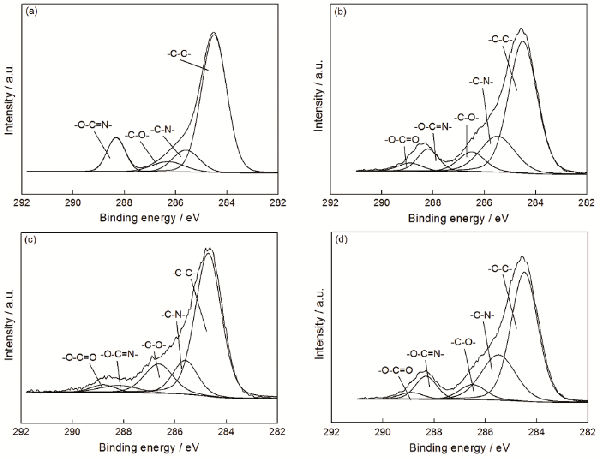
|
Treatment time/s |
Concentration of correlative functional groups/% | ||||
|---|---|---|---|---|---|
| -C-C- | -C-N- | -C-O- | -O-C=N- | -O-C=O | |
| 0 | 70.53 | 11.25 | 6.32 | 11.89 | 0 |
| 12 | 60.54 | 20.21 | 8.48 | 7.61 | 3.16 |
| 24 | 52.98 | 22.02 | 14.61 | 5.36 | 5.04 |
| 36 | 63.34 | 19.56 | 7.33 | 5.21 | 4.56 |


| Treatment time/s | Ra/nm | Rq/nm |
|---|---|---|
| 0 | 192.4 | 213.5 |
| 12 | 232.5 | 245.6 |
| 24 | 291.7 | 312.1 |
| 36 | 374.6 | 401.3 |
| Treatment time/s |
Single fiber tensile strength /MPa |
Standard deviation /MPa |
Decreasing rate/% |
|---|---|---|---|
| 0 | 5624 | 358 | 0 |
| 12 | 5444 | 425 | 3.2% |
| 24 | 5352 | 403 | 4.8% |
| 36 | 4988 | 416 | 11.3% |
 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0

 中冶有色技術(shù)平臺
中冶有色技術(shù)平臺交流會")

 2024年12月27日 ~ 29日
2024年12月27日 ~ 29日 合材料應(yīng)用技術(shù)創(chuàng)新論壇") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 廢水處理與資源化利用交流會") 2025年01月03日 ~ 05日
2025年01月03日 ~ 05日 大會") 2025年03月25日 ~ 27日
2025年03月25日 ~ 27日 資源科技創(chuàng)新發(fā)展論壇") 2025年04月27日 ~ 29日
2025年04月27日 ~ 29日 











































































