全部
位置:中冶有色 >
有色技術頻道 >
礦山技術
冶金技術
材料制備及加工技術
環(huán)境保護技術
分析檢測技術
功能材料技術
復合材料技術
新能源材料技術
合金材料技術
加工技術
> 加工技術
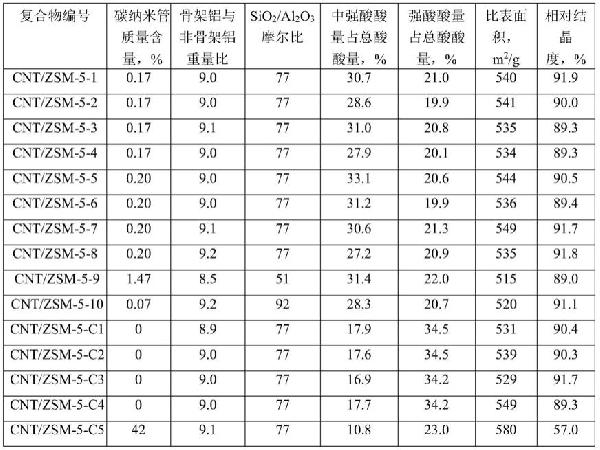
> 碳納米管/ZSM-5分子篩復合物及其合成方法和應用與流程
標題:碳納米管/ZSM-5分子篩復合物及其合成方法和應用與流程
 494
編輯:中冶有色技術網(wǎng)
來源:中國石油化工股份有限公司上海石油化工研究院
494
編輯:中冶有色技術網(wǎng)
來源:中國石油化工股份有限公司上海石油化工研究院
 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0









































































 中冶有色技術平臺
中冶有色技術平臺

 2024年12月27日 ~ 29日
2024年12月27日 ~ 29日 新論壇")
同創(chuàng)新發(fā)展論壇")

資源科技創(chuàng)新發(fā)展論壇")




